







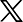

노스웨스턴大와 고정확도 머신러닝 모델 개발
전통적 고온 실험 없이 소재 개발 가능성 증명
전통적 고온 실험 없이 소재 개발 가능성 증명
|
한국과학기술원(KAIST)은 신소재공학과 홍승범 교수 연구팀이 미국 노스웨스턴대 크리스 울버튼 교수팀과 국제 공동연구를 통해,밀도범함수이론(DFT) 기반의 형성에너지 데이터를 활용해 합금이 녹을 때 성분이 유지되는지를 예측하는 고정확도 머신러닝 모델을 개발했다고 14일 밝혔다.
연구팀은 밀도범함수이론을 통해 계산한 형성에너지와 기존 실험적 융해 반응 데이터를 머신러닝에 결합해 4536개의 이원계 화합물에 대한 융해 반응 유형을 학습한 후 예측 모델을 구성했다.
다양한 머신러닝 알고리즘 중 특히 'XGBoost' 기반 분류 모델이 합금이 잘 섞이는지 여부에 대해 가장 높은 정확도인 82.5%의 예측 정확도를 달성했다.
또한 연구팀은 샤플리 기법(AI의 판단을 설명하는 도구)을 활용해 모델의 주요 특징들을 분석했다. 그중 기울기 변화가 크다는 것은 그 조성에서 에너지적으로 매우 유리한 상태가 형성된다는 뜻으로 '형성에너지 곡선의 기울기 변화'가 가장 중요한 인자로 도출됐다.
KAIST는 이번 연구의 가장 큰 의의로 고온 실험 없이도 소재의 융해 반응 경향성을 예측할 수 있다는 점을 꼽았다. 이는 고엔트로피 합금이나 초내열 합금 등 실험이 어려운 소재 군에서 매우 유용하며, 향후 복잡한 다성분계 합금 설계에도 확장될 수 있다.
또한 AI 모델이 도출한 주요 물리량은 합금이 잘 변하고 안정적인지 등에 대한 실제 실험 결과와 높은 일치도를 보였고, 향후 다양한 금속재료 개발 및 구조 안정성 예측 등에 널리 활용될 수 있을 것으로 기대된다.
홍승범 교수는 "이번 연구는 계산과 실험 데이터, 그리고 머신러닝의 융합을 통해 기존의 경험적 합금 설계 방식에서 벗어나 데이터 기반의 예측적 소재 개발이 가능하다는 가능성을 보여준 사례"라며 "향후 생성형 모델, 강화학습 등의 최신 AI 기술을 접목하면 완전히 새로운 합금을 자동으로 설계하는 시대가 열릴 것"이라고 말했다.






